2.2 Assembling a genome
When the human genome is 3 billion base pairs long, assembling short sequencing reads into a full genome is a major computational challenge.
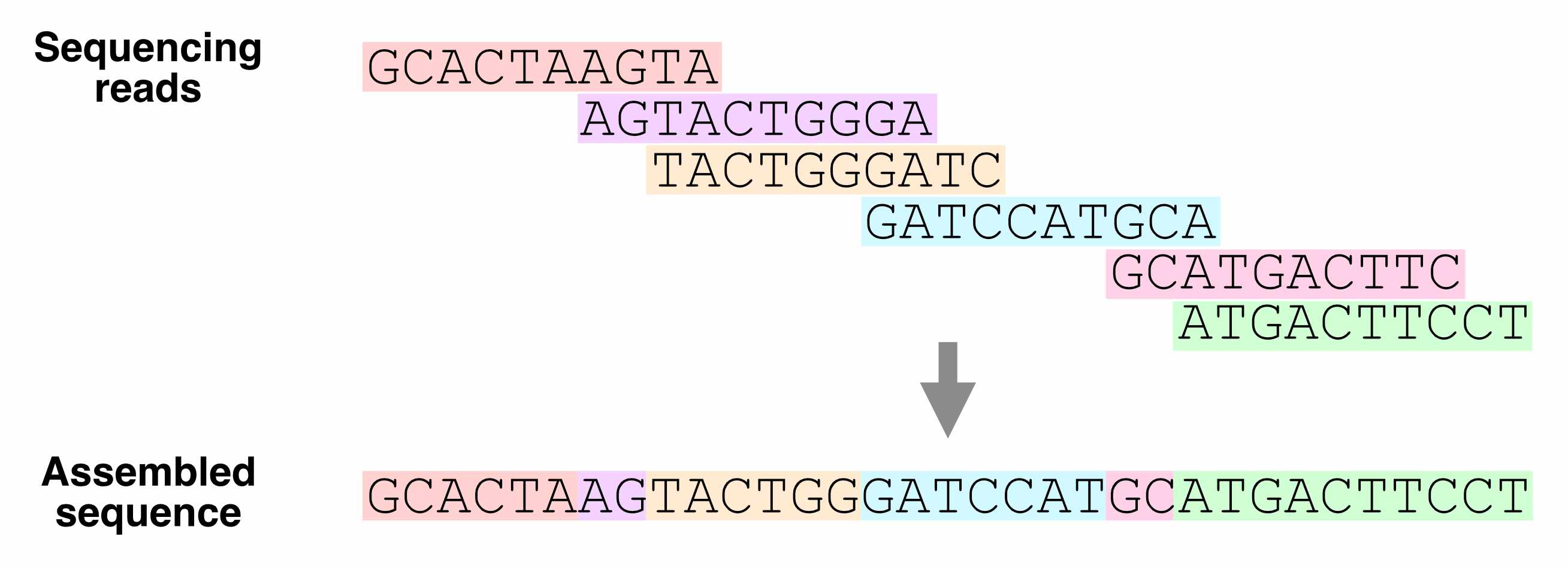
How is genome assembly performed?
We can combine sequencing reads that partially overlap with each other into longer sequences.
Which regions of the genome are hardest to assemble?
Ideally, with enough sequencing data, we would be able to reconstruct an entire genome from overlapping reads. In practice, genome assembly is complicated by repetitive DNA – sequences in different regions of the genome that are completely or nearly identical.
These repeats make it difficult (or impossible) to determine the order of the sequences around them, or how many copies of the repeat there are.
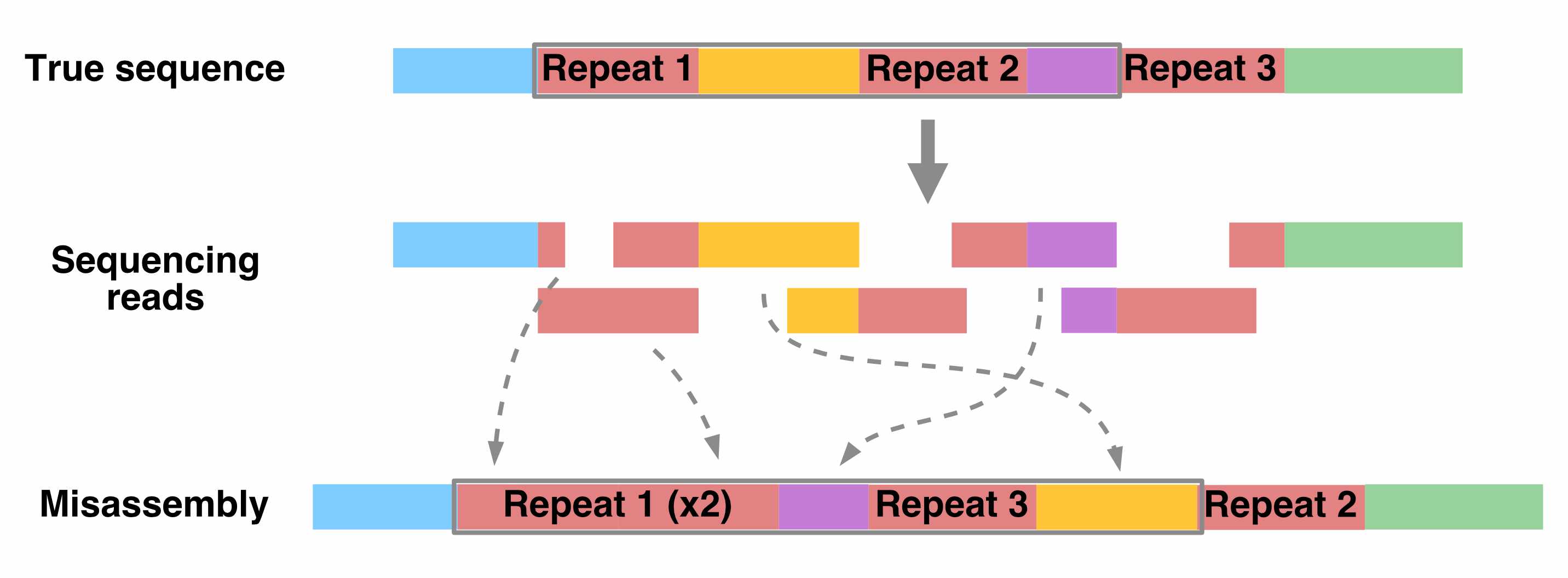
Resolving repetitive regions requires sequencing reads that are longer than the repeat itself, which allow us to determine the flanking sequences on the sides of the repeat.
Using such long-read sequencing technology (i.e., PacBio and Nanopore sequencing), the Telomere-to-Telomere consortium was able to create a complete, ungapped assembly of the human genome in 2021.