12.10 Building a phylogenetic tree
Using the distance matrix, we can now:
- Build a neighbor joining tree using the
nj()function - Use
HQ166910as the outgroup to root the tree (with theroot()function) - Use the
ladderize()function to re-orient the tree into a tidier format for plotting
# build a neighbor joining tree
tree <- nj(D)
# manually "root" the tree by setting HQ166910 as an outgroup
tree <- root(tree, which(ids == "HQ166910"))
# rotate tree at nodes to make it look tidier (i.e., "ladderized")
tree <- ladderize(tree)
# plot the tree
ggtree(tree) +
theme_tree2() +
geom_tiplab(label = names, size = 4) +
xlim(0, 1.2)
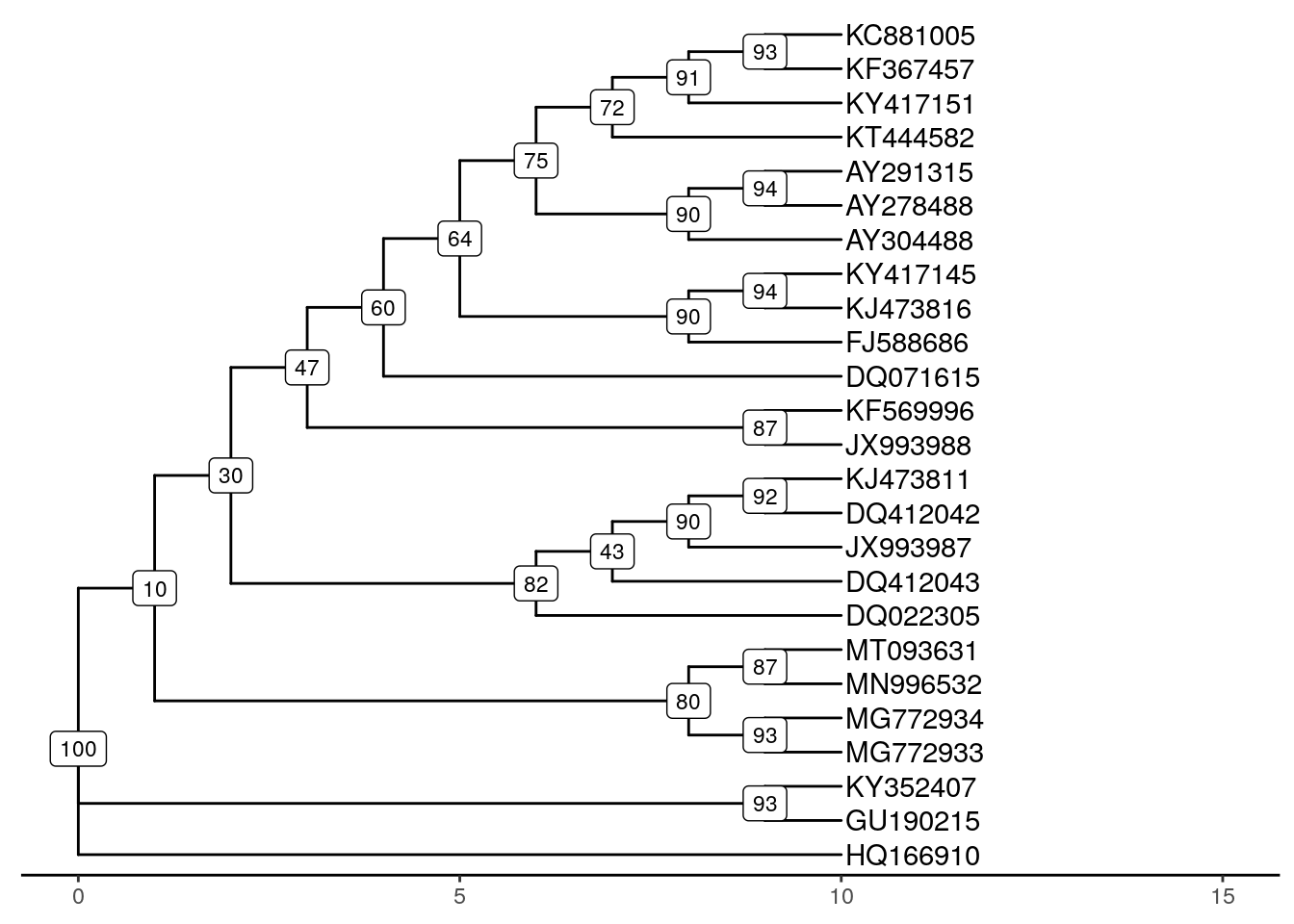
On the tree, we can see that the 2019-nCov sample (MT093631.2 Severe acute respiratory syndrome coronavirus 2) groups most closely with Bat coronavirus RaTG13.
Do you think this similarity is sufficient to confirm a bat origin of SARS-CoV-2?
Although the distance between SARS-CoV-2 and RaTG13 in the phylogeny looks small, it’s a large distance in phylogenetic space. Without sampling more deeply within intermediate strains between RaTG13 and SARS-CoV-2, we don’t know whether it passed through other mammalian species before being transmitted to humans.
yper ## Assess bootstrap support
A useful tool for evaluating confidence in a phylogenetic tree (or any other metric) is bootstrapping. This statistical method is based on resampling data with replacement from the original dataset.
In our case, we resample aligned sites (i.e., bases) from the original alignment, then build a new tree with the resampled data. By repeating this procedure many times, we can evaluate confidence in various parts of the original tree by asking how often the trees from resampled data contain these features.
Run the code below to implement bootstrapping in the boot.phylo() function. The output is a vector of bootstrap support values, which we can overlay onto the tree.
# set random seed
set.seed(123)
# bootstrap and build new trees to evaluate uncertainty
myBoots <- boot.phylo(tree, dna,
function(x) ladderize(root(nj(dist.dna(x,
model = "TN93")),
which(ids == "HQ166910"))),
rooted = TRUE)## Running bootstraps: 100 / 100
## Calculating bootstrap values... done.# replace "NA" with zero in bootstrap results; do not label terminal nodes
myBoots[is.na(myBoots)] <- 0
myBoots <- c(rep(NA, 25), myBoots)
# re-plot tree with bootstrap values
ggtree(tree, branch.length = "none") +
theme_tree2() +
geom_tiplab(label = names) +
geom_label(aes(label = myBoots), size = 3) +
xlim(0, 15)## Warning: Removed 25 rows containing missing values or values outside the scale range
## (`geom_label()`).