8.20 Homework
8.20.0.2 Assignment
Run a GWAS of \(\mathrm{IC_{50}}\) for the drug CB1908, using the same genotype data as before. The phenotypes are located in CB1908_IC50.txt.
Make a QQ plot and a Manhattan plot of your results. Do you have any genome-wide significant hits? Are they located in or near a gene? For the top GWAS hit, plot the phenotype stratified by genotype. (Use top_snp_hw.vcf to get the genotypes of the top hit.)
Solution
# perform association test with PLINK
system(command = "./plink --file genotypes --linear --allow-no-sex --pheno CB1908_IC50.txt --pheno-name CB1908_IC50")# read in gwas results
results <- read.table(file = "plink.assoc.linear", header = TRUE) %>%
mutate(index = row_number()) %>%
arrange(P)
# qq plot
qq(results$P)
# manhattan plot
manhattan(results)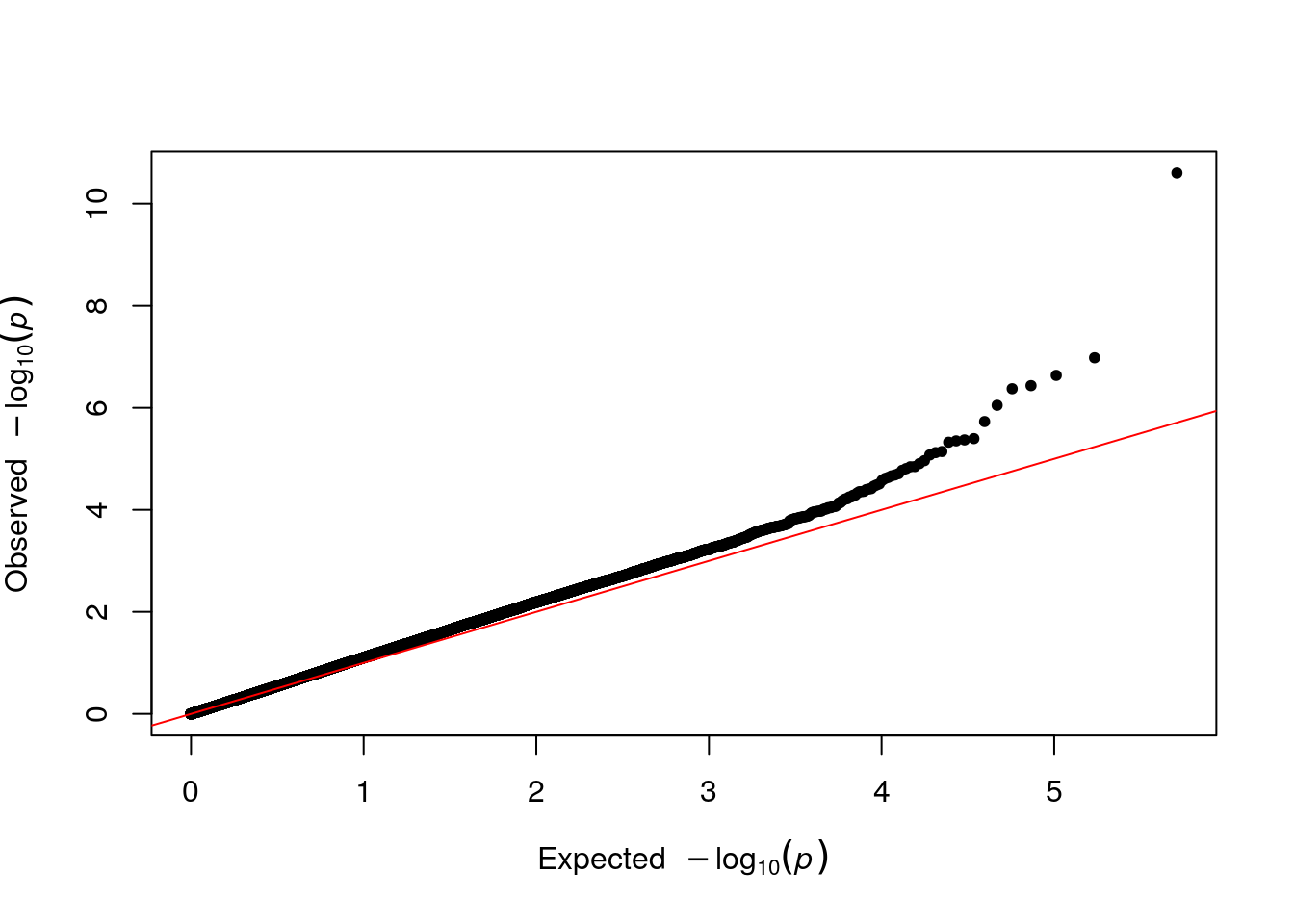
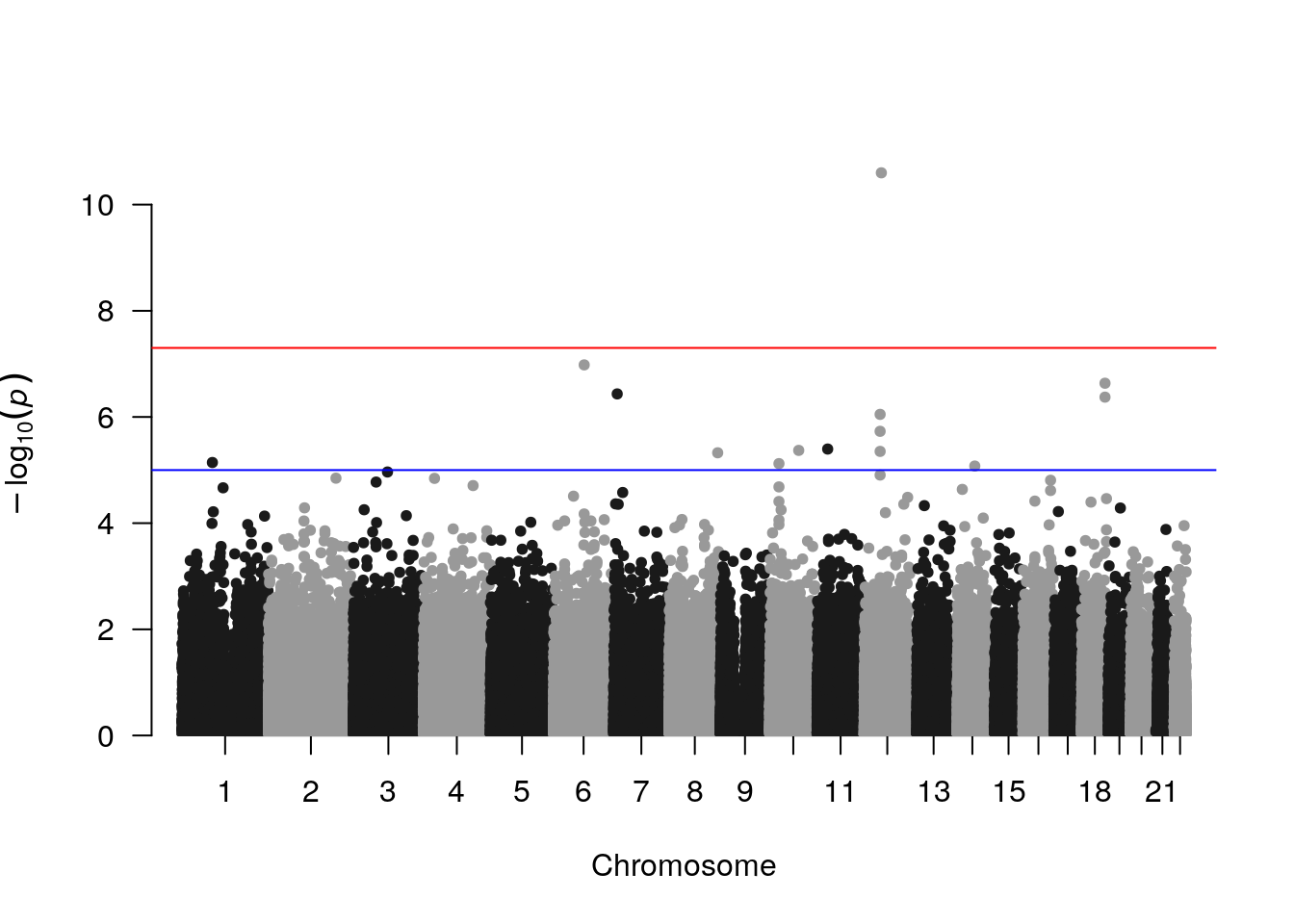
On the Manhattan plot, there’s one hit that reaches genome-wide significance.
## CHR SNP BP A1 TEST NMISS BETA STAT P
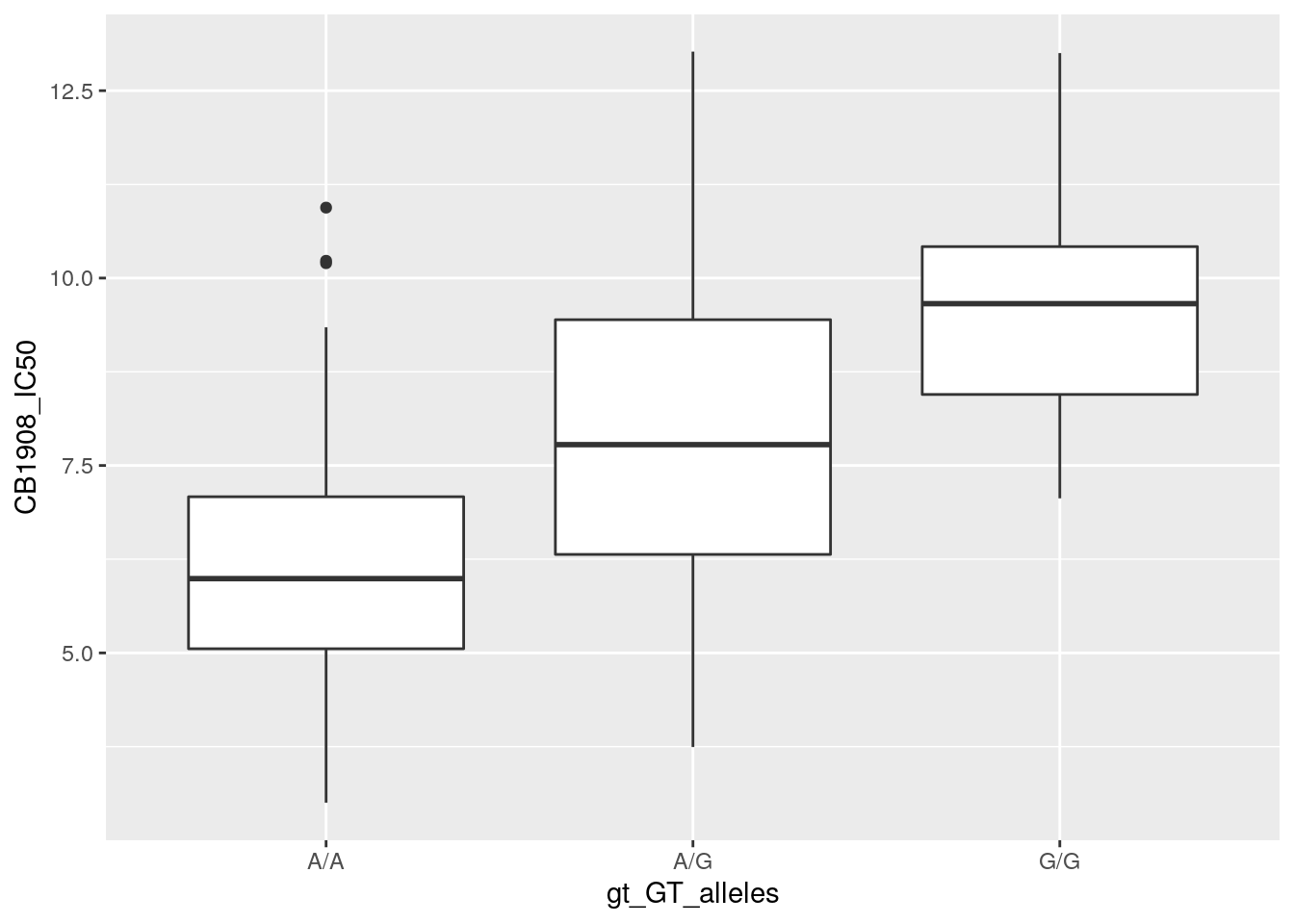
## 1 12 rs10876043 49190411 G ADD 161 1.779 7.18 2.518e-11From looking it up in the UCSC Genome Browser, rs10876043 lies within an intron of the DIP2B gene. Finally, we plot this SNP’s genotype stratified by phenotype, using top_snp_hw.vcf.
## Scanning file to determine attributes.
## File attributes:
## meta lines: 27
## header_line: 28
## variant count: 1
## column count: 185
## Meta line 27 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 1
## Character matrix gt cols: 185
## skip: 0
## nrows: 1
## row_num: 0
## Processed variant: 1
## All variants processed## Extracting gt element GT# get genotype dataframe
top_snp_gt <- top_snp$gt %>%
drop_na()
# read in phenotype dataframe
phenotypes <- read.table("CB1908_IC50.txt", header = TRUE)
# merge genotype and phenotype info
gwas_data <- merge(top_snp_gt, phenotypes,
by.x = "Indiv", by.y = "IID")
# plot genotype by phenotype boxplots
ggplot(data = gwas_data) +
geom_boxplot(aes(x = gt_GT_alleles,
y = CB1908_IC50))## Warning: Removed 2 rows containing non-finite outside the scale range
## (`stat_boxplot()`).