12.12 Homework
12.12.0.2 Assignment
hw_accessions.txt and hw_aligned.fa provide the alignments and metadata for 24 human SARS-CoV-2 sequences, plus the bat RaTG13 coronavirus strain.
Use this data to repeat the analysis we did in class (compute distance matrix, construct neighbor joining tree, perform bootstrapping for confidence values) to build a phylogeny of SARS-CoV-2 strains. You should use RaTG13 (accession: MN996532) as an outgroup.
Solution
Read in accession data:
accessions <- read.table("hw_accessions.txt", sep = "\t", header = TRUE)
# make vectors of accessions IDs and full names
ids <- accessions$id
names <- accessions$nameConstruct distance matrix:
# read fasta file
dna <- read.dna("hw_aligned.fa", format = "fasta", as.matrix = TRUE)
# compute pairwise distance matrix
D <- dist.dna(dna, model = "TN93", as.matrix = TRUE)Build neighbor joining tree:
# make the tree
tree <- nj(D)
# manually root tree at RaTG13 coronavirus
tree <- root(tree, which(ids == "MN996532"))
# ladderize tree
tree <- ladderize(tree)Bootstrap to determine tree uncertainty:
set.seed(123)
myBoots <- boot.phylo(tree, dna,
function(x) ladderize(root(nj(dist.dna(x,
model = "TN93")),
which(ids == "MN996532"))),
rooted = TRUE)## Running bootstraps: 100 / 100
## Calculating bootstrap values... done.Plot the bootstrapped tree:
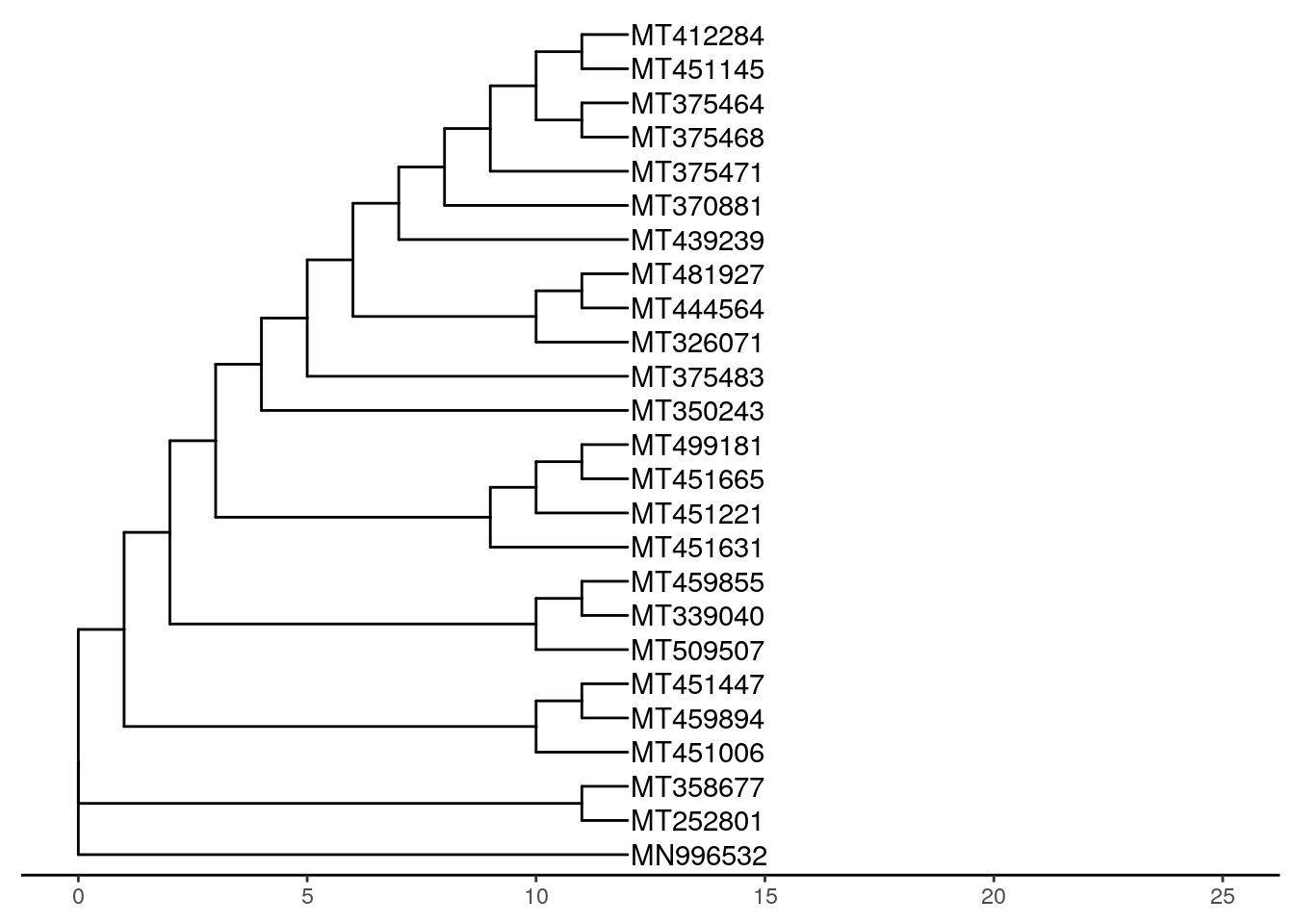
ggtree(tree, branch.length = "none") +
theme_tree2() +
geom_tiplab(label = names) +
geom_nodelab(label = myBoots, geom = "label", fill = "#deebf7") +
xlim(0, 25)