8.18 Top GWAS SNP
One common future direction for GWAS studies is following up on the top SNP(s). Read in top_snp.vcf, a VCF of just the top SNP in the dataset, so that we can plot boxplots of the top SNP genotype stratified by phenotype:
## Scanning file to determine attributes.
## File attributes:
## meta lines: 27
## header_line: 28
## variant count: 1
## column count: 185
## Meta line 27 read in.
## All meta lines processed.
## gt matrix initialized.
## Character matrix gt created.
## Character matrix gt rows: 1
## Character matrix gt cols: 185
## skip: 0
## nrows: 1
## row_num: 0
## Processed variant: 1
## All variants processed## Extracting gt element GTtop_snp_gt <- top_snp$gt %>%
drop_na()
# merge with phenotype data
gwas_data <- merge(top_snp_gt, phenotypes,
by.x = "Indiv", by.y = "IID")
# plot boxplots
ggplot(data = gwas_data) +
geom_boxplot(aes(x = gt_GT_alleles,
y = GS451_IC50))## Warning: Removed 1 row containing non-finite outside the scale range
## (`stat_boxplot()`).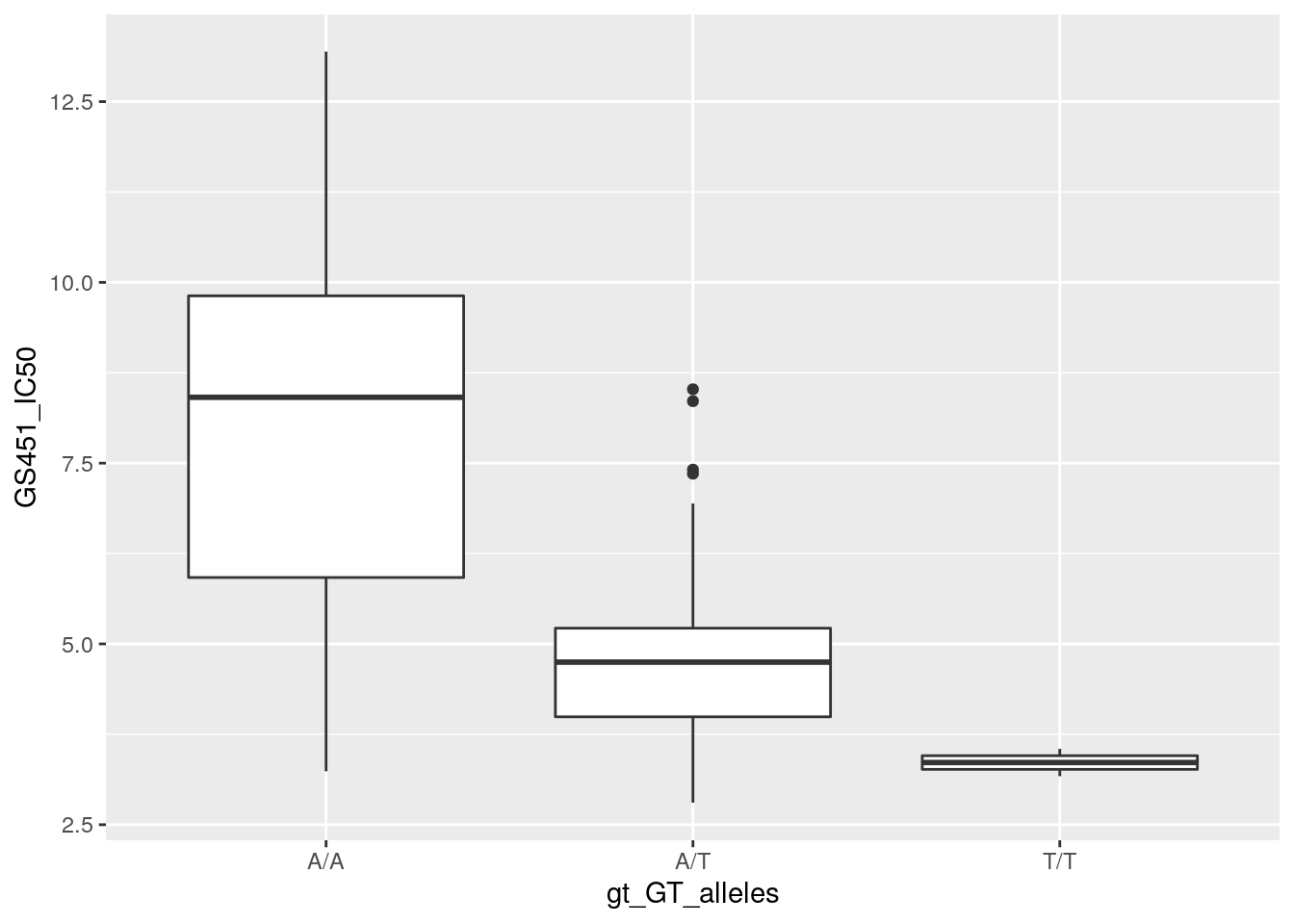
Other potential follow-up directions include:
- Investigating the genomic environment in the UCSC Genome Browser
- Looking at nearby haplotype structure with LDproxy
- Note that the genotype data we’re using come from the Yoruba population
- Using the Geography of Genetic Variants browser to find the global allele frequencies of the variant
- Search for SNP in a phenotype database to see if there are other associations with it