12.8 Neighbor joining trees
One approach to building a phylogeny of sequences is neighbor joining, which clusters sequences based on their pairwise genetic distance. In this approach, we:
- Start with a star phylogeny that assumes all samples are equally related (Fig. 5A)
- Compute a pairwise distance matrix between sequences, and look for the pair of sequences that are most similar to each other
- We join these two sequences to form a new node (Fig. 5B)
- The distance matrix is re-computed and this process repeats until all nodes are joined (Fig. 5C)
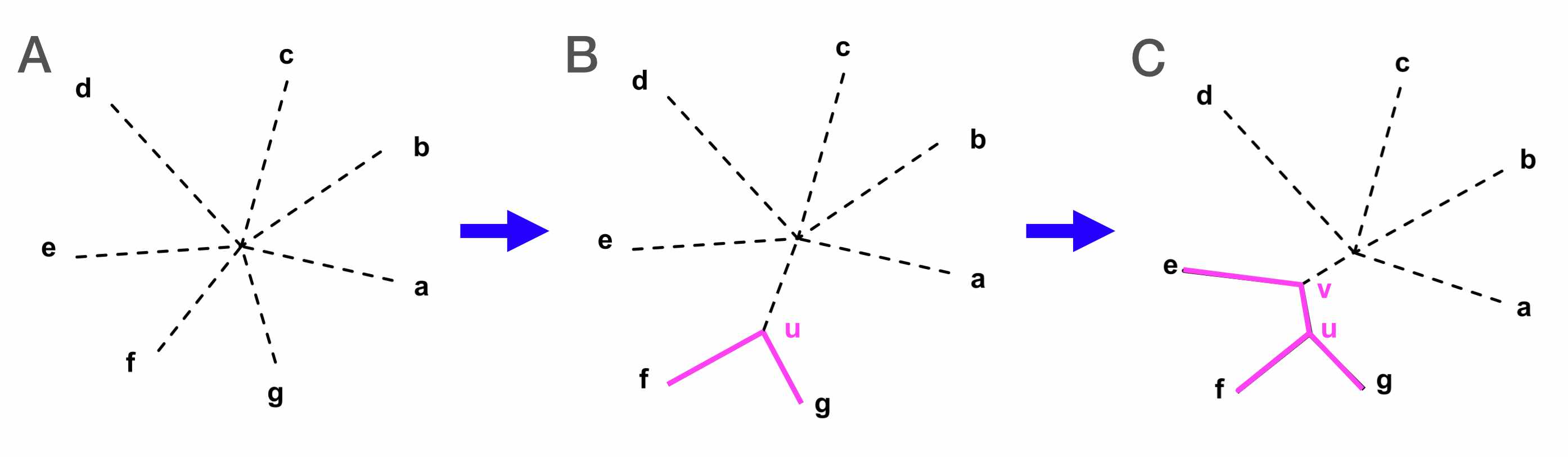
Fig. 5. Steps for constructing a neighbor joining tree (source).