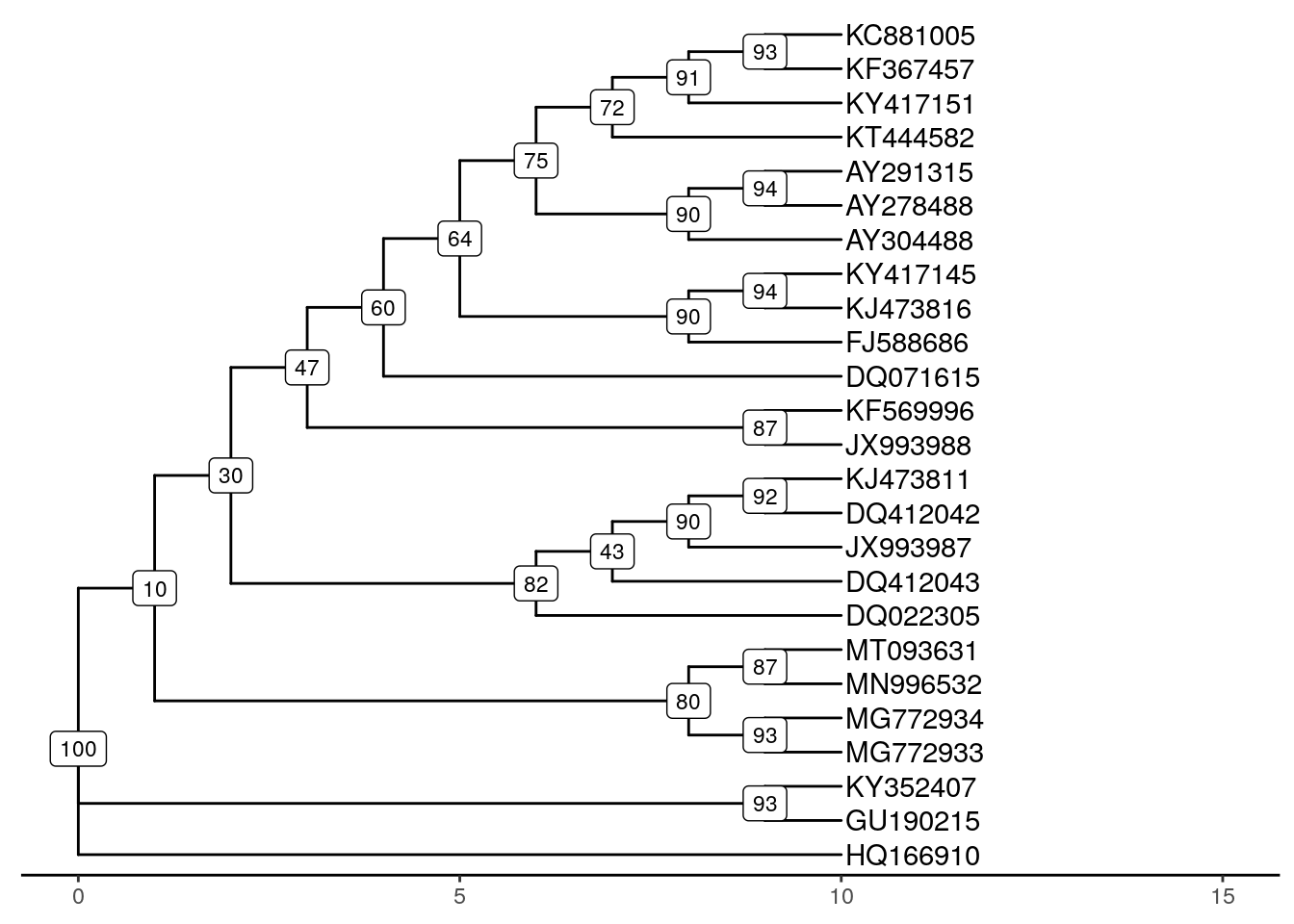
12.11 Assess bootstrap support
A useful tool for evaluating confidence in a phylogenetic tree (or any other metric) is bootstrapping. This statistical method is based on resampling data with replacement from the original dataset.
In our case, we resample aligned sites (i.e., bases) from the original alignment, then build a new tree with the resampled data. By repeating this procedure many times, we can evaluate confidence in various parts of the original tree by asking how often the trees from resampled data contain these features.
Run the code below to implement bootstrapping in the boot.phylo() function. The output is a vector of bootstrap support values, which we can overlay onto the tree.
# set random seed
set.seed(123)
# bootstrap and build new trees to evaluate uncertainty
myBoots <- boot.phylo(tree, dna,
function(x) ladderize(root(nj(dist.dna(x,
model = "TN93")),
which(ids == "HQ166910"))),
rooted = TRUE)##
Running bootstraps: 100 / 100
## Calculating bootstrap values... done.# replace "NA" with zero in bootstrap results; do not label terminal nodes
myBoots[is.na(myBoots)] <- 0
myBoots <- c(rep(NA, 25), myBoots)
# re-plot tree with bootstrap values
ggtree(tree, branch.length = "none") +
theme_tree2() +
geom_tiplab(label = names) +
geom_label(aes(label = myBoots), size = 3) +
xlim(0, 15)## Warning: Removed 25 rows containing missing values (geom_label).